A vaccine against HIV is on the horizon after scientists showed a new drug triggered a protective immune response in humans and stopped two thirds of monkeys becoming infected. In the 35 years since the HIV epidemic began, just four vaccines have been tested on humans, with the best only lowering infection rates by 31 per cent, leading to trials being discontinued. But an international team of scientists showed that the new vaccine boosted the immune systems of nearly 400 healthy adults. And when vaccinated rhesus monkeys were exposed to the disease six times, only one third became infected.
AIDS & HIV
 In early 1981 doctors began to see patients with strange symptoms in hospitals across New York City and San Francisco. Young men were showing signs of rare diseases that were very unusual for their age. In July 1981 the New York Times reported the outbreak of a rare form of cancer specifically among gay men called Kaposi's SarcomaANCHOR. Over the next months and years doctors saw increasing numbers of gay men who were having serious difficulties in fighting off a number of common infections and cancers.
In early 1981 doctors began to see patients with strange symptoms in hospitals across New York City and San Francisco. Young men were showing signs of rare diseases that were very unusual for their age. In July 1981 the New York Times reported the outbreak of a rare form of cancer specifically among gay men called Kaposi's SarcomaANCHOR. Over the next months and years doctors saw increasing numbers of gay men who were having serious difficulties in fighting off a number of common infections and cancers.
Doctors soon discovered a commonality among these men: they were lacking CD4+ cells which are essential for maintaining a healthy immune system. One year later the Centers for Disease Control had enough evidence to make the link between the illness and blood, and they named the disease AIDS (Acquired Immune Deficiency Syndrome)ANCHOR. Over the next few years it became evident that the cause of AIDS was infectious, but it wasn't until 1984 that a virus named HIV (Human Immunodeficiency Virus) was shown to be the cause of AIDS.
To read more about this history of HIV and AIDS see the history pages on the AVERT website, which includes a timeline which highlights some of the most important developments.
What is HIV?
Testing for HIV
Animal models of HIV
Development and use of anti-retroviral medication
The search for a vaccine
Approaches to vaccine development
The STEP and Phambili MRK-Ad5 vaccine trials
Examples of HIV research using animals
The way forward
References
What is HIV?
The HIV retrovirus is an RNA based virus. When HIV infects cells in the body its viral RNA is recognized by the cell which then produces the complementary DNA strands. Over time this HIV DNA becomes integrated into the DNA of the host cell to become a provirus. From this point on whenever the cell replicates the HIV DNA is also replicated. The cell can remain dormant for a long time, but when it is activated it produces new viral particles which go on to infect other cells.
This chain reaction leads to the body initiating an immune response which includes the production of antibodies which are specific to the HIV virus. Whilst infection with HIV immediately begins to infect and damage cells in the body there is a long asymptomatic period (average of 10 years) between infection and the development of any symptoms. During this time the level of the virus in the blood drops to a low level, but people remain infectious.
Over time however the virus targets cells in the immune system which leaves the body susceptible to infections. The HIV virus specifically targets CD4+ T cells which are important components of the human immune systemANCHOR. The virus attaches itself to the outside of the T-cells before infecting them. This leads to apoptosis and the immune system becomes progressively weaker until the patient picks up an infection or cancer and is considered to be suffering from AIDSANCHOR. It is important to note that people do not die of AIDS; they die from the other conditions, illnesses and infections that their bodies can no longer fight off due to their weakened immune systems.
Françoise Barré-Sinoussi and Luc Montagnier were awarded a share of the 2008 Nobel Prize in Physiology or Medicine for their discovery of HIV as a cause of AIDS. Their identification of HIV as a lentivirus meant that scientists had some idea of its behaviour based on past lentivirus research on animals. The fast identification of the virus allowed health officials to advise the public on how to prevent transmission and ultimately reduced the spread of the virus.
Testing for HIV
 The fact that the immune response to the HIV virus can be slow means that it can take up to six months for the body to produce significant quantities of antibodies. This made the initial search for a test difficult.
The fact that the immune response to the HIV virus can be slow means that it can take up to six months for the body to produce significant quantities of antibodies. This made the initial search for a test difficult.
The transmission of HIV to chimpanzees in the lab gave the first animal model of HIV. This was crucial to the development of blood tests allowing both diagnosis and the screening of blood donations. The test uses a technique commonly used in other areas of immunology which can detect the presence of a specific antibody. Called ELISA, it involves taking diluted blood serum and applying it to a plate with antigens attached. The test is sensitive enough to detect whether the blood contains HIV antibodies (which bind to the specific HIV antigens on the plate) even when they are at low levels. The ELISA method quickly became accepted worldwide as an effective way to screen for HIV. A negative ELISA test is sufficient to show that HIV is not present in a sample, but a second test is used to confirm a positive result from the ELISA test.
Whilst most people produce enough HIV antibodies to be detected by the ELISA test within 6 to 12 weeks, it can sometimes (in rare cases) take up to 6 months. Because of this 'window period' between infection and the production of antibodies there is the risk of the test showing a false negative result. Therefore it is recommended that a person who thinks they may have been infected with HIV is tested twice, once three months after exposure, and again after around six months.
Animal models of HIV
BLT mouse
Bone marrow-Liver-Thymus (BLT) mice have become an important part of research into new treatments and vaccinesANCHOR. With bone marrow, Thanks to animal testing involving liver and thymus tissue transplants from humans, the mice can be regarded as having a human immune systemANCHOR. This allows them to become infected by HIV when introduced vaginally and exhibit many of the hallmarks of the disease in humans.
SIV as a model of HIV
Simian Immunodeficiency Virus (SIV) is similar to HIV and both are particularly prone to mutation, so single individuals will pick up variations in the RNA structure of the HIV they carry over their lifetime. The amino acid structure of an individual HIV-1 protein has around 95% similarity with the same viral protein isolated from another individual.
HIV-1 shares the highest degree of similarity (85%) with the chimpanzee strain of SIV. It is believed that HIV-1 mutated from this strain of SIV before jumping species to infect humans and HIV-1 can infect chimpanzees. Although there have been instances of chimpanzees developing AIDSANCHOR, these are very rare, and since the early 1990s scientists have moved away from the chimpanzee as a model of HIV infection.
It was soon discovered that macaques are susceptible to SIV, and that the virus goes on to cause a fatal immunodeficiency syndrome in this speciesANCHOR. Researchers have also found that SIV is sensitive to similar drugs to HIV and they have exploited these similarities to develop and test many antiviral medications, particularly those used in prophylaxis.
A disadvantage with SIV is that the sequence of its genes and proteins differ in places to that of HIV, which means that some HIV vaccines cannot be tested directly against SIV. In the mid-1990s, scientists overcame this difficulty and developed SHIVs: genetically engineered viruses consisting of a SIV backbone and the HIV genes Env, Rev, Tat, and Vpu8. While early strains of SHIV failed to lead to AIDS in macaques, pathogenic strains such as SHIV 89.6p were eventually producedANCHOR.
HIV and SIV both infect cells through their interaction with the CD4 protein found on the surface of some T-cells, but infection also requires the virus to interact with another protein called a co-receptor. In most cases of early HIV-1 and SIV infection the co-receptor used is CCR5, but in other cases another co-receptor named CXCR4 is usedANCHOR. Several of the SIV strains, and more importantly SHIV strains such as SHIV 89.6p, also use CXCR4 as their co-receptor - a difference that turned out to be vital for future HIV vaccine development.
Use and development of anti-retroviral medication
Control of HIV in patients
Whilst there is currently no cure for AIDS, there are treatments which aim to slow the progression from infection with HIV to AIDS. Retroviral therapies, usually used in combination, have led to a reduction in deaths associated with HIV infection in the developed world.
1986 saw the first clinical trial of an anti-retroviral medicine called AZT more commonly known as Retrovir and Retrovis. AZT had been studied for retroviral research in mice before the AIDS epidemic began, and experiments had shown it to have activity against retroviruses at doses which were non-toxic in animalsANCHOR. The activity of AZT against HIV was soon confirmed in animals, and in 1987 it became the first anti-retroviral to be licensed by the FDA as a treatment for AIDS and HIV. Soon other anti-retroviral drugs followed, which were more effective and less toxic than AZT. Unfortunately the virus quickly developed resistance to all of these compounds which is why treatment with combinations of drugs is now the norm.
 A major advance came with the development of drugs that inhibit the activity of HIV-1 protease, an enzyme that is necessary for the formation of virus particles. Saquinavir entered clinical trials in 1991, and in 1995 became the first protease inhibitor to be approved by the FDA. During pre-clinical testing, in vitro studies had shown it to have potent anti-HIV activity, while animal tests indicated that it had an acceptable toxicity profile and that clinically relevant doses could be achieved by oral dosingANCHOR. The publication of the 3-D structure of the HIV-protease protein guided the development of Indinavir which proved to be more potent than Saquinavir. Over 150 lead and intermediate compounds were tested during the development of Indinavir, and once again studies of oral bioavailability and toxicity in animals played a key role in identifying the compound that was taken into clinical trialsANCHOR. Indinavir quickly became a standard component of highly active antiretroviral therapy (HAART) that has greatly reduced AIDS related mortality in developed countries.
A major advance came with the development of drugs that inhibit the activity of HIV-1 protease, an enzyme that is necessary for the formation of virus particles. Saquinavir entered clinical trials in 1991, and in 1995 became the first protease inhibitor to be approved by the FDA. During pre-clinical testing, in vitro studies had shown it to have potent anti-HIV activity, while animal tests indicated that it had an acceptable toxicity profile and that clinically relevant doses could be achieved by oral dosingANCHOR. The publication of the 3-D structure of the HIV-protease protein guided the development of Indinavir which proved to be more potent than Saquinavir. Over 150 lead and intermediate compounds were tested during the development of Indinavir, and once again studies of oral bioavailability and toxicity in animals played a key role in identifying the compound that was taken into clinical trialsANCHOR. Indinavir quickly became a standard component of highly active antiretroviral therapy (HAART) that has greatly reduced AIDS related mortality in developed countries.
By 1996 the combination therapy, consisting of cocktails of anti-virals taken together, had increased life expectancy enormously. While these drugs are not a cure, and need to be taken for life, the development of around 20 anti-virals which are effective against HIV is considered one of the great medical successes of the late 20th Century. A study in 2013 in South Africa showed that HIV patients have a life expectancy of 80% of the general population, provided that they are given antiretrovirals at an early enough stageANCHOR.
Pre-exposure prophylaxis
Pre-exposure prophylaxis (PrEP) is a course of anti-retroviral treatment aimed at people with a high risk of contracting HIV. This includes men who have sex with men, couples where only one partner is HIV-positive and people who are unable to insist on condom use.
Truvada, a combination treatment of tenofovir and emtricitabine, was approved in the US in 2012 for people in these high risk groups. This treatment was previously tested in macaque monkeys and in BLT mice, where it showed strong protection against rectally-introduced SHIV and vaginally-introduced HIV respectivelyANCHOR,ANCHOR.
Results from a phase III large-scale clinical trial, iPrEx, published in 2010 demonstrated that Truvada provided an additional 44 per cent protection against contracting HIV among gay men. The level of protection was higher among those that reliably took the pills dailyANCHOR.
Preventing transmission
The HPTN 052 trial from the HIV Prevention Trials Network studied the use of antiretroviral drugs given to HIV positive patients as a way to prevent transmission to uninfected partners in heterosexual relationships. The results, revealed in 2011, showed a 96% reduction in transmission. This was far higher than expected and the trial was hailed as ‘Breakthrough of the Year’ by Science magazineANCHOR. Antiretroviral drugs are known to reduce the amount of virus present in patients’ bloodstreams and so it was expected that they would also reduce transmission. However, with no precise figures on this effect, public health officials could not advocate their use in this fashion with the risk of people reducing the use of condoms.
The search for a vaccine
The need for a vaccine to protect populations is clear, and difficulties in controlling the spread of HIV, particularly in sub-Saharan Africa, combined with the high cost of anti-retroviral drugs, have driven the need for an HIV vaccine.
 Despite the optimism that a vaccine would be possible in the early days of HIV researchANCHOR, it was soon realised that developing a vaccine would pose more technical difficulties than anticipated. The problem is that the HIV virus is unlike any virus studied before, because its genetic material is encoded in RNA the virus is able to mutate rapidly. This characteristic helps it to evade the body's immune response, as antibodies are only able to recognise specific regions of the viral surface. As vaccines work by stimulating the production of antibodies, this is a significant problem. The viral envelope of HIV is also very flexible, and changes shape as it attaches to T-cells, making it difficult for antibodies to bind to the virus and stop the process. However, the past 25 years of research have given several leads, especially thanks to animal experiments, as to ways to overcome these problems and produce an effective HIV vaccine. One such lead relies on the fact that there are regions of the virus which are very stable and do not tend to mutate.
Despite the optimism that a vaccine would be possible in the early days of HIV researchANCHOR, it was soon realised that developing a vaccine would pose more technical difficulties than anticipated. The problem is that the HIV virus is unlike any virus studied before, because its genetic material is encoded in RNA the virus is able to mutate rapidly. This characteristic helps it to evade the body's immune response, as antibodies are only able to recognise specific regions of the viral surface. As vaccines work by stimulating the production of antibodies, this is a significant problem. The viral envelope of HIV is also very flexible, and changes shape as it attaches to T-cells, making it difficult for antibodies to bind to the virus and stop the process. However, the past 25 years of research have given several leads, especially thanks to animal experiments, as to ways to overcome these problems and produce an effective HIV vaccine. One such lead relies on the fact that there are regions of the virus which are very stable and do not tend to mutate.
Evidence that human antibodies can neutralise HIV in animal modelsANCHOR , the fact that some people have immune responses which are able to control the virus for years without developing AIDS, and the demonstration that some vaccines are effective against SIV in macaquesANCHOR indicate that developing an effective vaccine is possible.
Approaches to vaccine development
Live-attenuated virus
Many approaches have been investigated in the search for a successful HIV vaccine. One approach is to develop a Live-attenuated vaccine, which involve the use of forms of the live virus which have been altered, either genetically, by mutating specific points on the RNA, or by other means, rendering them unable to cause disease. This type of vaccine gives a closer simulation of natural HIV infection than inactivated viruses, and generates good immune responses. Attenuated SIV vaccines have had mixed success in protecting macaques against the disease in challenge studies, where they are exposed to SIV after inoculation. In some studies animals were successfully protectedANCHOR, but not in othersANCHOR. Unfortunately live-attenuated SIV vaccines themselves caused AIDS when given to infant macaques, and long-term follow up of adult macaques showed that here too the live vaccine sometimes eventually caused AIDSANCHOR, making this approach unsuitable for human trials. Disappointing as this was, it did at least provide further evidence that it was possible to stimulate an immune response that could prevent SIV/HIV infection.
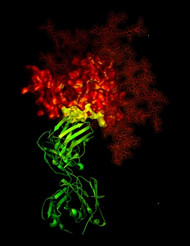
X-ray crystallographic image showing the broadly neutralizing antibody b12 (green ribbon) in contact with a critical target (yellow) for vaccine developers on HIV-1 gp120 (red). © NIH
Viral envelope proteins
With live-attenuated vaccines appearing unpromising a variety of other vaccination techniques have been studiedANCHOR. The classical approach to vaccine development is to use an inactivated form of the virus or part of the virus, such as the viral envelope protein, to induce a humoral immune response, generating antibodies which will recognise the virus and prevent an infection from establishing itself in the body. The outer envelope protein of the virus, gp120, was an obvious target and a purified form was developed to be used as a vaccine. AIDSVAX, an early gp120 subunit vaccine, was able to block low-dose infection in chimpanzees by the atypical CXCR4-binding HIV strain HIV-1 IIIB, and was taken into human clinical trials. Unfortunately AIDSVAX failed to protect in the clinical trialsANCHOR, a failure that was predicted by its earlier failure to prevent infection of macaques by a pathogenic CCR5-binding SIV strain. Efforts to develop subunit-based vaccines have continued, but the results against CCR5-binding SIV strains have mostly been disappointing, with at best partial protection against homologous SIV strainsANCHOR.
DNA vaccines
Live recombinant, or DNA, vaccines have also been developed, in which another virus is used to transfer genes from HIV or SIV into cells. The infection in these cells can then be 'recognised' by the immune system, leading to an immune response. In contrast with the humoral response stimulated by subunit vaccines, live-recombinant vaccines stimulate cytotoxic T- cells, a type of lymphocyte which lacks the CD4 receptor HIV uses to enter the cell, and so cannot be infected by HIVANCHOR. The normal role of these cells in the body is to control a viral infection and kill virus-infected cells before they divide. There is good evidence that these cells control HIV during the first stages of infection, preventing the development of AIDS, often for many years. Animal studies in macaques have shown that the ability of vaccines to stimulate cytotoxic T-cells has an impact on the spread of the infection, reducing viral load. Cytotoxic T-cells only attack cells which contain the virus, so they do not prevent infection, but they do reduce the impact of the virus on the immune system, and a low viral load can prevent transmission, reducing the spread of disease.
The STEP and Phambili MRK-Ad5 vaccine trials
The MRK-Ad5 vaccine was viewed as a promising advance in vaccine development and its progress was watched closely. After going through the development process, using non-animal and animal-based tests, and early clinical trials it was withdrawn from phase IIb clinical trials in 2007ANCHOR.
The MRK-Ad5 vaccine consisted of 3 HIV genes, carried into the body using a common cold virus, adenovirus-5, as a vector. The key HIV genes carried by the virus were hoped to be sufficient to give immunity to HIV, allowing the body's T-cells to identify and kill cells containing the virus, without either the HIV genes or the adenovirus which carried them being able to cause disease.
Though it did not stimulate production of protective antibodies it had stimulated cytotoxic T-cells strongly in animal studies, and it was hoped that it would therefore reduce viral load in people who became infected with HIV, and perhaps even lower infection rates. The second set of phase II trials were designed to test how effective the vaccine would be. Two separate trials were run, one, known as STEP, took place in America and Australia, while a separate trial, 'Phambili', took place in South Africa.
Initial analysis of the STEP study showed that of 741 people who received a single dose of the vaccine 24 became infected with HIV. This was compared with 762 people who received the placebo, of whom 21 became infected with HIV. Although this result was not statistically significant, further analysis was carried out, and an increased likelihood of infection was found in relation to a particular group of male volunteers.
Male volunteers who already had strong immunity to adenovirus-5 prior to vaccination seemed more likely to become infected with HIV once they had received the vaccine (21 out of 392 men who had high levels of antibody to adenovirus, 9 out of 386 in the control group). Similar vaccines that use different adenoviruses are now in developmentANCHOR, so it is important to understand the issues behind the MRK-Ad5 vaccine.
Later analysis of the two trials suggested that the vaccine seemed to raise the risk of infection by 41%. However, some scientists have questioned the extent of the effect as it included HIV infections after the trial was unblinded. This raises the risk that those who discovered that they had received the vaccine would have been more likely to engage in risky sexual behaviourANCHOR.
The MRK-Ad5 vaccine was fully developed, and extensively studied in non-human primates before human trials began. Macaques were given a vaccine similar to MRK-Ad5, and were then infected with SHIV 89.6p, a hybrid SIV, genetically engineered to contain genes from HIV. While monkeys which received the vaccine were not protected against infection with SHIV they did have lower viral loads than those which did not receive the vaccine and did not develop AIDS.
The decision to proceed to clinical trials with MRK-Ad5 was criticized at the time by scientists who raised concerns about the reliability of SHIV 89.6p as a model of HIV infection, since the manner in which it infects cells of the immune system differs from that seen with HIV infectionANCHOR. Most importantly MRK-Ad5 failed to prevent infection or have an impact on disease progression in macaques exposed to SIVMAC239, a strain of SIV that uses CCR5 and whose course of infection closely matches that seen in the majority of HIV casesANCHOR,ANCHOR.
Examples of HIV research using animals
19/02/15 A new gene therapy vaccine seems to completely protect monkeys from HIV
A new approach to vaccination seems to completely protect monkeys from HIV. Vaccines usually prepare the immune system to fight an infection. Instead, scientists have altered the DNA of monkeys to give their cells HIV-fighting properties. This technique uses gene therapy to introduce a new section of DNA – containing the instruction to build tools to neutralize HIV - inside healthy muscle cells. The monkeys who received the injection were protected from all types of HIV for at least 34 weeks. Researchers want to start human trials as fast as possible and they believe that this approach may be useful in people who already have HIV.
http://www.bbc.co.uk/news/health-31511244
27/02/15 Llamas are immune to HIV - new hope for a vaccine
Llamas might be the new hope for a new AIDS vaccine or treatment – llamas appear to be immune to HIV. Llama antibodies, which develop in response to the virus potently neutralizes more than 95% of HIV strains. In humans, the antibodies are completely ineffective at halting the virus they have evolved to target. Unlike human antibodies, llama antibodies have a single chain of proteins, which allows them to accurately aim at specific viruses compared to a more scatter-gun approach to the human immune system, attacking all foreign viruses.
http://www.usatoday.com/…/lima-peru-llama-aids-hiv/23884381/
27/02/15 Llamas are immune to HIV - new hope for a vaccine
Llamas might be the new hope for a new AIDS vaccine or treatment – llamas appear to be immune to HIV. Llama antibodies, which develop in response to the virus potently neutralizes more than 95% of HIV strains. In humans, the antibodies are completely ineffective at halting the virus they have evolved to target. Unlike human antibodies, llama antibodies have a single chain of proteins, which allows them to accurately aim at specific viruses compared to a more scatter-gun approach to the human immune system, attacking all foreign viruses.
http://www.usatoday.com/…/lima-peru-llama-aids-hiv/23884381/
03/07/15 New AIDS vaccine protects 50% of monkeys against SIV
A new AIDS vaccine protects Monkeys against SIV. With a 50% success rate in monkeys with SIV, the vaccine is already being trialled in humans. The researchers hope that the vaccine will work better in humans than in monkeys, given that the monkeys had been given a gigantic dose of SIV, much more than average people get in an average sexual exposure to HIV. HIV vaccines have been proven many times unsuccessful because the virus infects the same cell the body uses to fight against the infection, it changes a lot so becomes hard to recognise and the human body doesn’t seem to create powerful broadly neutralizing antibodies against the virus. This new vaccine uses a common cold virus called adenovirus 26 that activates an immune response. Then a second vaccine is given with bits of HIV attached. The immune system cells will also "see" the attached bit of HIV and, the researchers hope, react against any HIV virus should the vaccinated person ever be exposed.
http://www.nbcnews.com/health/health-news/new-aids-vaccine-protects-monkeys-n385751
21/01/16 A protein found in primates and humans may stop HIV progression
A protein found in both primates and humans may stop HIV progression and switch on the immune system, according to a new study published January 14 in the journal, Heliyon. TRIM5α from the rhesus macaque (TRIM5αRh) is a restriction factor that shows strong activity against HIV-1
http://www.alnmag.com/news/2016/01/protein-part-treats-hiv-infections-non-human-primates
http://www.heliyon.com/article/e00056/
21/03/16 Giving antibodies to infant macaques exposed to HIV-like virus could clear the infection
An infant rhesus macaques treated with antibodies within 24 hours of being exposed to SHIV (simian HIV) was completely cleared of the virus researchers at Oregon National Primate Research Center found. SHIV-infected non-human primates can transmit SHIV to their offspring through milk feeding, just as humans can transmit HIV from mother to child through breastfeeding and during childbirth. In humans, a combination of measures for mothers and infants, including antiretroviral therapy (ART), C-section delivery and formula feeding, have decreased the rate of mother-to-child HIV transmission from 25% to less than 2% since 1994. However, despite this decrease, approximately 200,000 children are infected with HIV each year worldwide, primarily in developing countries where ART is not readily available. Whilst it is recommended to treat human babies with ART during the last month of gestation, the few days after delivery, and during breastfeeding time frames, risks still remain, including toxicities associated with long-term ART use, the development of drug-resistant viral variants, and lack of access to prenatal care prior to delivery. This discovery indicates that using new methods, such as antibodies, to limit infection after exposure in newborns could be advantageous.
http://www.ohsu.edu/xd/about/news_events/news/2016/03-21-giving-antibodies-to-inf.cfm
19/05/16 HIV DNA removed from infected rats and mice
Gene-editing technology has allowed scientists to remove HIV DNA from various organs of infected mice and rats. This research could lead to an outright cure. A similar study involving a larger group of animals will be conducted before clinical trials in humans can take place.
http://www.independent.co.uk/news/science/hiv-aids-cure-virus-disease-dna-genes-temple-university-research-gene-editing-a7037571.html
22/07/16 Researchers used mice to show that SIV can transmit from chimps to human cells
Certain strains of the Simian Immunodeficiency Virus (SIV) are able to infect humans. This evidence suggests that the disease which affects chimpanzees may be the original source of HIV in humans. It is believed that humans who ate infected bush meat was the source of the disease. The researchers used humanised mice and found the SIV strains were able to infect them easily.
14/10/16 Promising HIV therapy
A new therapy for HIV shows promise in monkeys. A combination of drugs was able to protect rhesus macaques for two years from SIV - the monkey (simian) version of HIV. The combination included standard antiretroviral drugs and combined it with an experimental antibody.
http://www.independent.co.uk/news/science/hiv-cure-breakthrough-monkeys-humans-vedolizumab-primate-a7361146.html
22/09/17 New antibody attacks 99% of HIV strains
Scientists have developed an antibody that attacks 99 per cent of HIV strains. Already tested on monkeys, trials on human beings could begin next year. Up until now, HIV has been difficult to treat because of its ability to mutate and change its appearance. However, the study, published in the journal Science, combines three broadly neutralising antibodies capable of targetting HIV viruses, into an even more powerful "tri-specific antibody" that attacks three key parts of the virus. This molecule is more potent and has greater breadth than any single naturally occurring antibody that's been discovered. This makes it harder for the virus to evade the antibodies. Experiments on 24 monkeys showed none of those given the tri-specific antibody developed an infection when they were later injected with the virus.
http://www.bbc.com/news/health-41351159
06/07/18 HIV vaccine on horizon as jab triggers immunity in humans and stops monkeys being infected
The way forward
There is now a broad recognition within the HIV vaccine research community that faced with understandable pressure to get candidate vaccines into clinical trials, and disappointing results from tests performed against stringent SIV strains, the bar for progression to human clinical trials was set too low. Among several recommendationsANCHOR,ANCHOR,ANCHOR there is general agreement that before proceeding to clinical trials a candidate vaccine should:
– Demonstrate efficacy against stringent challenge viruses such as SIVMAC239 and SIVMAC251
– Demonstrate efficiency in rhesus monkeys that lack MHC alleles associated with efficient virologic control (Mamu-A*01, Mamu-B*17, Mamu-B*08)
– Demonstrate efficacy against both homologous and non-homologous SIV challenge
– Target virus effectively during early infection of mucosal tissues
– Induce both an antibody mediated and T-cell mediated immune response
There is still an urgent need for new treatments to control HIV, and it is clear that more basic research needs to be done. One approach that has been studied extensively for the past decade is the prime-boost strategy, which involves immunization with a DNA vaccine followed a few months later by a boost immunization with a subunit based vaccine, with the objective of stimulating a stronger immune response that involves both the antibody and T-cell arms of the immune system.
References
- http://www.nytimes.com/1981/07/03/us/rare-cancer-seen-in-41-homosexuals.html
- 80 Days That Changed the World: A Name for the Plague July 27, 1982 http://content.time.com/time/specials/packages/article/0,28804,1977881_1977895_1978703,00.html
- Garg H, Mohl J, Joshi A (2012) HIV-1 induced bystander apoptosis Viruses 4(11):3020-43 doi: 10.3390/v4113020
- Cummkins NW and Badley AD (2010) Mechanisms of HIV-associated lymphocyte apoptosis: 2010 Cell Death and Disease 1, e99 doi: 10.1038/cddis.2010.77
- Shultz LD et al (2012) Humanized mice for immune system investigation: progress, promise and challenges Nat Rev Immunol. 12(11):786–798 doi:10.1038/nri3311
- Melkus MW et al (2006) Humanized mice mount specific adaptive and innate immune responses to EBV and TSST-1 Nature Medicine 12(11):1316-22 doi:10.1038/nm1431
- Novembre, FJ et al (1997) Development of AIDS in a chimpanzee infected with human immunodeficiency virus type 1 J Virol 71(5):4086-4091
- Brenchley JM et al (2004) CD4+ T cell depletion during all stages of HIV disease occurs predominantly in the gastrointestinal tract J Exp Med. 200(6):749-759
- Feinberg MB and Moore JP (2002) AIDS vaccine models: Challenging challenge viruses Nature Medicine 8:207-210
- Feinberg MB and Moore JP (2002) AIDS vaccine models: Challenging challenge viruses Nature Medicine 8:207-210
- Mitsuya, H et al (1985) 3’-Azido-3’-deoxythymidine (BW A509U): an antiviral agent that inhibits the infectivity and cytopathic effect if human T-lymphotropic virus type III/ lymphadenopathy-associated virus in vitro, Proc. Natl. Acad. Sci. USA. 82,
- Duncan IN and Redshaw S (2001) Discovery and early development of Saquinavir, in Ogdon RC and Flexner CW ed, Protease inhibitors in AIDS therapy, Informa HealthCare, London, pages -48
- Dorsey BD et al. (1994) L-735,524: the design of a potent and orally bioavailable HIV protease inhibitor. J. Med. Chem., 37, 3443-3451.
- Johnson LF et al (2013) Life Expectancies of South African Adults Starting Antiretroviral Treatment: Collaborative Analysis of Cohort Studies PLoS Medicine 10(4):e1001418 doi:10.1371/journal.pmed.1001418
- Cohen MS and Kashuba AD (2008) Antiretroviral Therapy for Prevention of HIV Infection: New Clues From an Animal Model. PLoS Medicine 5(2):20 doi:10.1371/journal.pmed.0050030
- Denton PW et al (2008) Antiretroviral Pre-exposure Prophylaxis Prevents Vaginal Transmission of HIV-1 in Humanized BLT Mice. PLoS Medicine 5(1): e16 doi:10.1371/journal.pmed.0050016
- Grand RM et al (2010) Preexposure chemoprophylaxis for HIV prevention in men who have sex with men The New England Journal of Medicine 363:2587-2599 doi:10.1056/NEJMoa1011205
- http://www.sciencemag.org/content/334/6063/1628.full
- Statement issued 27th Feb 1986 by W. Dowdle, AIDS coordinator of the Public Health Service, Public Health Reports
- Gauduin MC et al (1997) Passive immunization with a human monoclonal antibody protects hu-PBL-SCID mice against challenge by primary isolates of HIV-1. Nat Med 3 (12) 1389-93.
- Hu SL et al (1992) Protection of macaques against SIV infection by subunit vaccines of SIV envelope glycoprotein gp160, Science 255 (5043) 456-459.
- Connor RI et al (1998). Temporal Analyses of Virus Replication, Immune Responses, and Efficacy in Rhesus Macaques Immunized with a Live, Attenuated Simian Immunodeficiency Virus Vaccine. J Virol, 72, 7501-7509.
- Lewis MG et al (1999) Limited Protection from a Pathogenic Chimeric Simian-Human Immunodeficiency Virus Challenge following immunization with Attenuated Simian Immunodeficiency Virus. J Virol, 73, 1262-1270.
- Baba, TW et al. (1999) Live attenuated, multiply deleted simian immunodeficiency viruscauses AIDS in infant and adult macaques. Nature Medicine 5:194–203.
- Girard MP, Osmanov SK, and Kieny MP (2006)A review of vaccine research and development: the human immunodeficiency virus (HIV) Vaccine 24(19):4062-4081.
- Brenchley JM et al (2004) CD4+ T cell depletion during all stages of HIV disease occurs predominantly in the gastrointestinal tract J Exp Med. 200(6):749-759
- Girard MP, Osmanov SK, and Kieny MP (2006)A review of vaccine research and development: the human immunodeficiency virus (HIV) Vaccine 24(19):4062-4081.
- Girard MP, Osmanov SK, and Kieny MP (2006)A review of vaccine research and development: the human immunodeficiency virus (HIV) Vaccine 24(19):4062-4081.
- http://www.hvtn.org/media/pr/step1207.html
- http://www.nature.com/news/hiv-vaccine-raised-infection-risk-1.13971
- http://www.nature.com/news/hiv-vaccine-raised-infection-risk-1.13971
- Veazey RS et al. (1998) Gastrointestinal tract as a major site for CD4+ T cell depletion and viral replication in SIV infection, Science, 280 427.
- Allen TM et al. (2002) Effects of cytotoxic T lymphocytes (CTL) directed against a single simian immunodeficiency virus (SIV) Gag CTL epitope on the course of SIVmac239 infection. J Virol. 76(20):10507-10511.
- Casimiro, DR et al. (2005)Attenuation of simian immunodeficiency virus SIVmac239 infection by prophylactic immunization with DNA and recombinant adenoviral vaccine vectors expressing Gag. J. Virol. 79:15547–15555.
- Morgan C et al. (2008) The use of nonhuman primate models in HIV vaccine development. PLoS Medicine 12;5(8):e173
- Barouch DH (2008) Challenges in the development of an HIV-1 vaccine. Nature 455(7213):613-619
- Hasse AT (2010) Targeting early infection to prevent HIV-1 mucosal transmission Nature 464(7286):217-223
Last edited: 24 January 2024 16:10